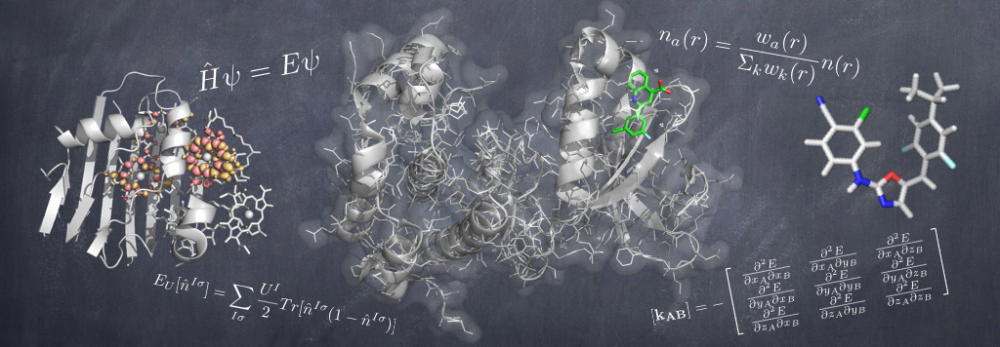
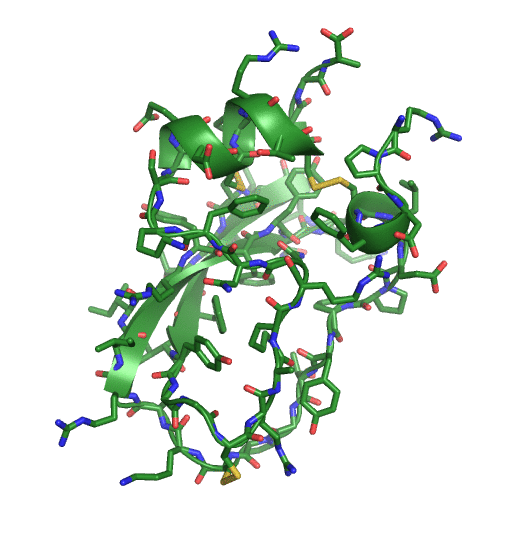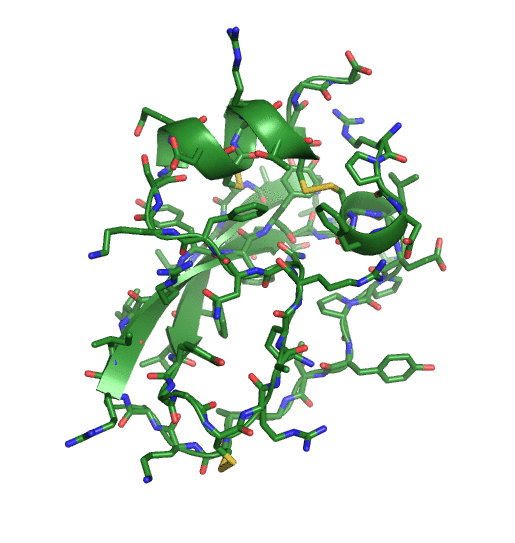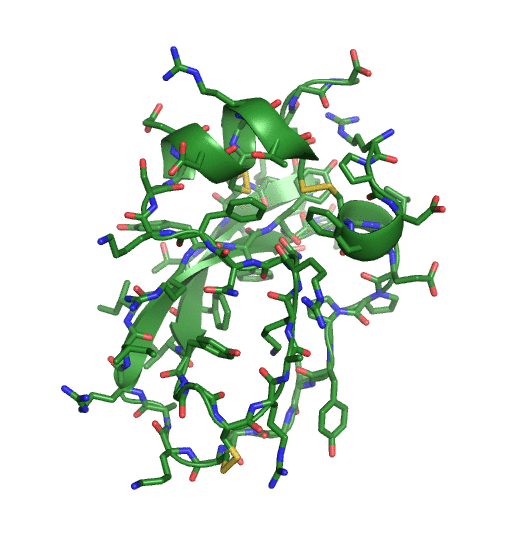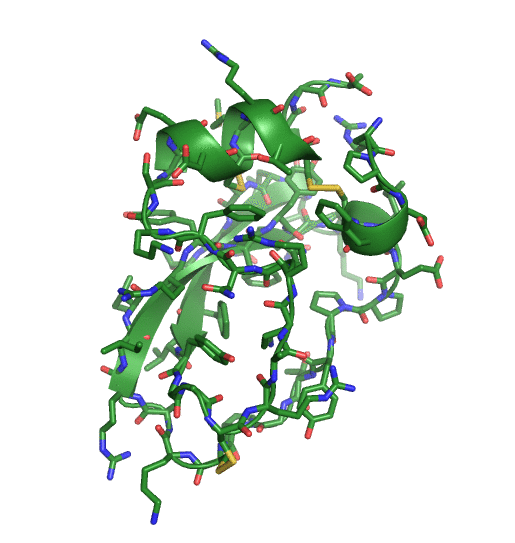
This page describes the goals, design and applications of the QUantum mechanical BEspoke (QUBE) force field model. A recent paper gives an overview of the methods and benchmarks the accuracy. Links to software are provided at the bottom of the page.
The QUBE Philosophy
{kind=link}
- Based on quantum mechanics. Where possible, all parameters of the QUBE force field are derived from quantum mechanical (QM) simulations, rather than fit to experiment. This substantially reduces the time-consuming and tedious fitting process.
- Bespoke. With the spread of low-cost computing resource, it is now feasible to reduce reliance on transferable libraries of parameters, and instead derive bespoke force fields directly for the system under study.
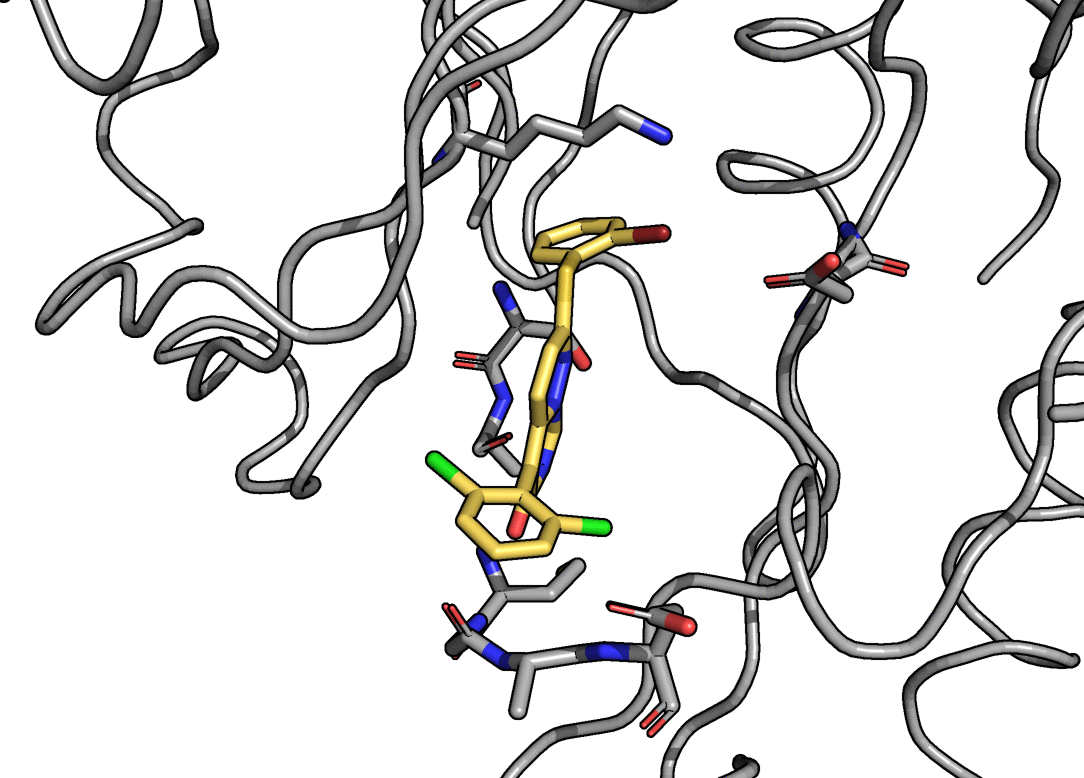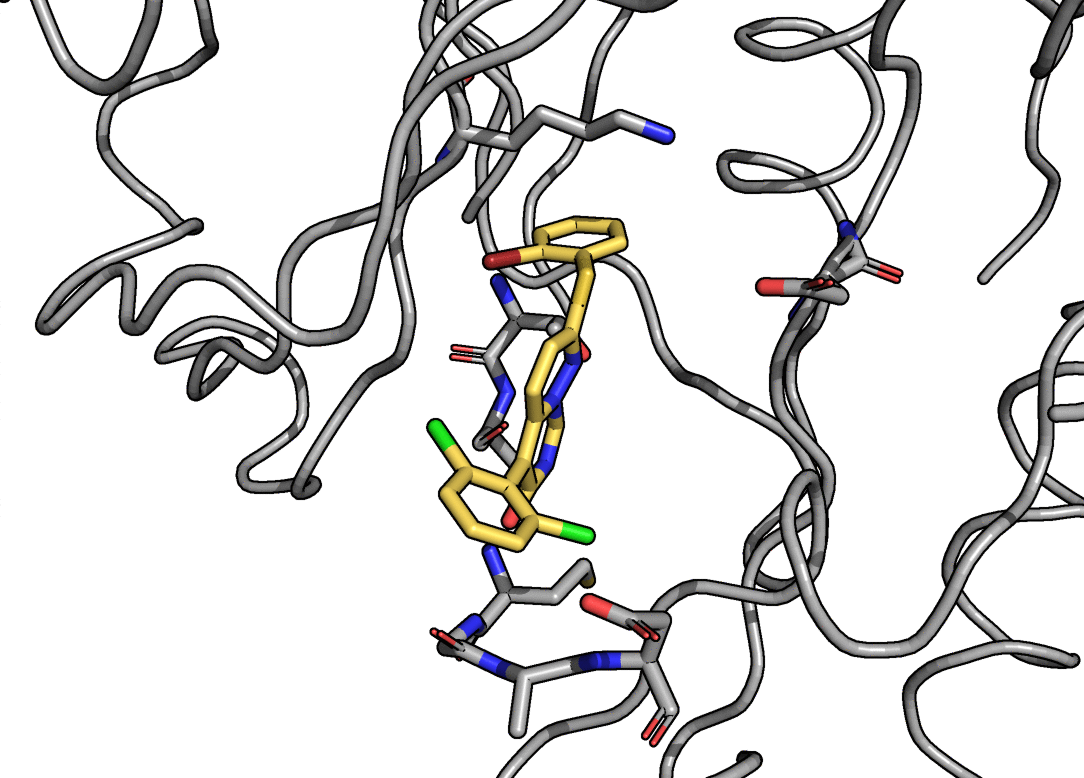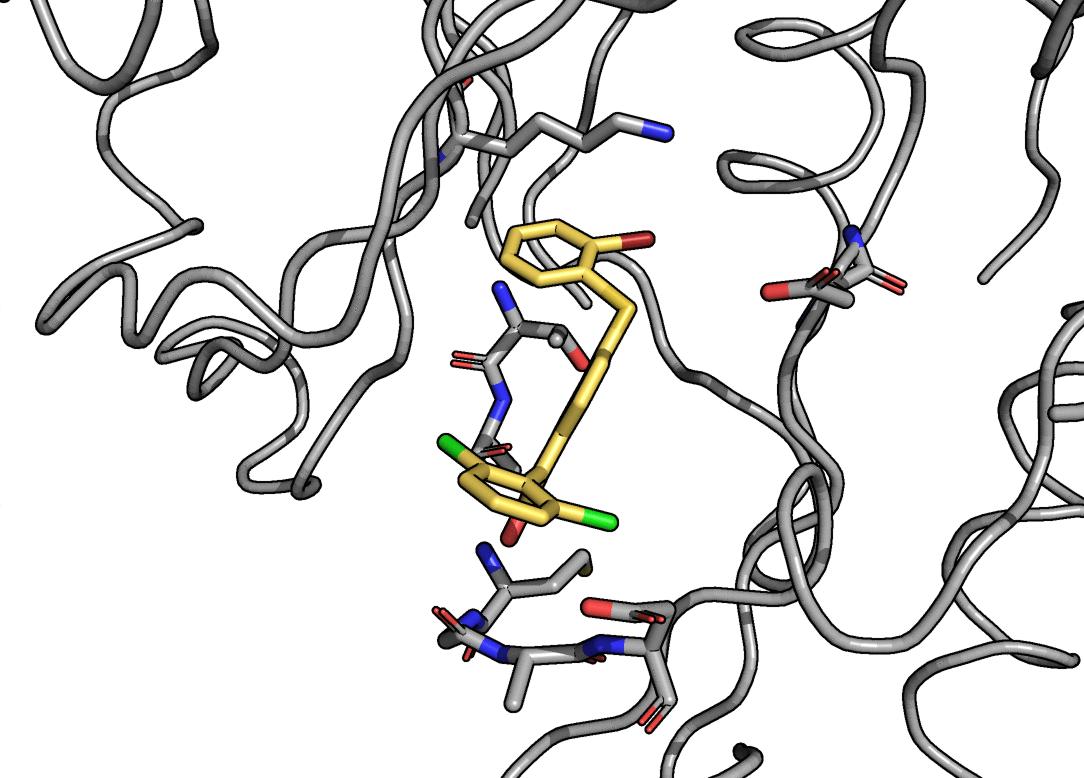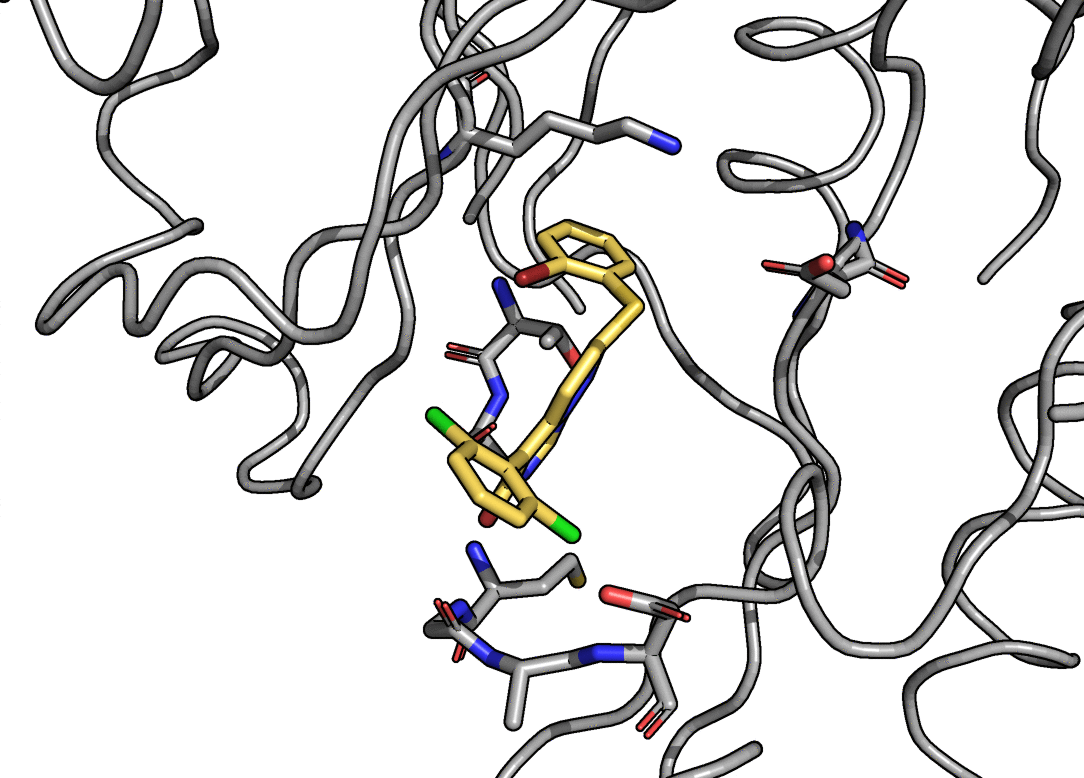
- Accurate. Ongoing work is benchmarking the accuracy of QUBE against experimental liquid properties, protein dynamics, and protein-ligand binding free energies.
- Scaling. By implementing our methods in the ONETEP linear-scaling density functional theory code, our non-bonded parameters can be derived for molecules containing thousands of atoms, including entire proteins.
- Automated and easy-to-use. This is a work in progress but see below for details of the QUBEKit software package for small molecule force field derivation, and QUBEKit-pro, our tool for setting up protein MD simulations using the QUBE force field.
- Wide range of applications. Although our main interest is in biomolecular force fields for computer-aided drug design, we are also investigating the use of QUBE for organic and organometallic optical materials.
How QUBE works
We have developed unique methods to ensure that our force field is, as much as possible, derived from quantum mechanics, rather than fit to experiment.
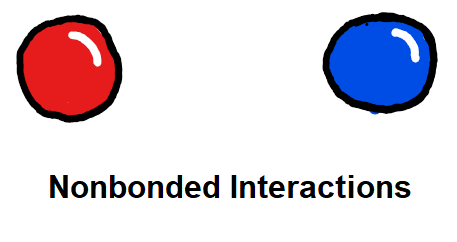
Charge and Lennard-Jones non-bonded parameters are derived from atoms-in-molecule partitioning of the quantum mechanical electron density. The developed methods significantly reduce the number of empirical parameters needed to construct molecular mechanics force fields, naturally include polarization effects in charge and Lennard-Jones parameters, and scale well to systems comprised of thousands of atoms, including entire proteins. The methods used are described in detail here.
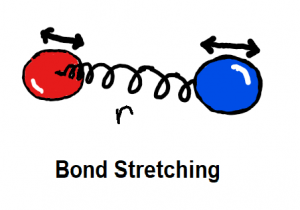
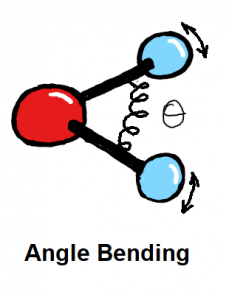
Bond stretching and angle bending are generally modelled as harmonic springs in MM force fields. In QUBE, harmonic bond and angle force constants are derived directly from the QM Hessian matrix using our modified Seminario method. QUBE reproduces QM vibrational frequencies to an accuracy of around 6%.
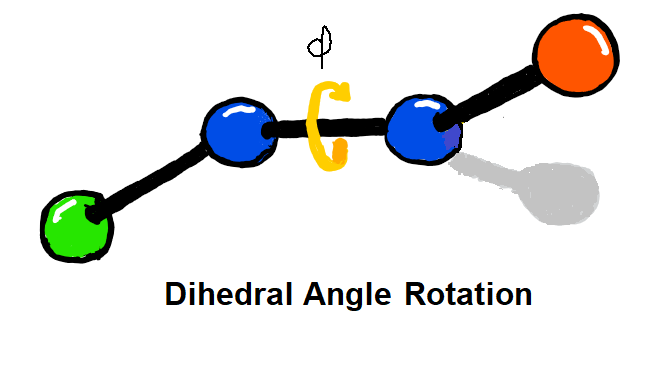
Dihedral parameters are fit to QM torsional scans using constrained geometry optimisation. Set-up of torsional scans and parameter fitting are both automated, and support for weighting and regularisation schemes are also provided.
QUBEKit
 The QUBEKit package automates the derivation of the QUBE bond, angle, dihedral and non-bonded force field parameters for small organic molecules. In the first version, QUBEKit required the BOSS molecular mechanics and Gaussian and ONETEP quantum mechanics codes for parameterisation, but support for alternative packages (e.g. psi4 and OpenMM) is now available. QUBEKit provides simulation-ready input files in BOSS, GROMACS, or OpenMM format.
The QUBEKit package automates the derivation of the QUBE bond, angle, dihedral and non-bonded force field parameters for small organic molecules. In the first version, QUBEKit required the BOSS molecular mechanics and Gaussian and ONETEP quantum mechanics codes for parameterisation, but support for alternative packages (e.g. psi4 and OpenMM) is now available. QUBEKit provides simulation-ready input files in BOSS, GROMACS, or OpenMM format.
![]()
This work is funded by the UKRI.
Image credits: Lauren Nelson.