Our group uses and contributes to a wide range of software packages for research in physics and chemistry, including:
- Open Force Field Initiative: an open approach to better force fields;
- OpenMM: A high-performance toolkit for molecular simulation;
- Sire: molecular simulation framework;
- MACE: fast and accurate machine learning interatomic potentials with higher order equivariant message passing;
- MCPRO: statistical mechanics of proteins in solution;
- ONETEP: linear-scaling density functional theory.
You can find a recent tutorial that was presented at the CCPBioSim annual training week, covering many of the concepts of force field assignment here.
Software developed in our research group:
For a complete and up-to-date list, see our group Github page.
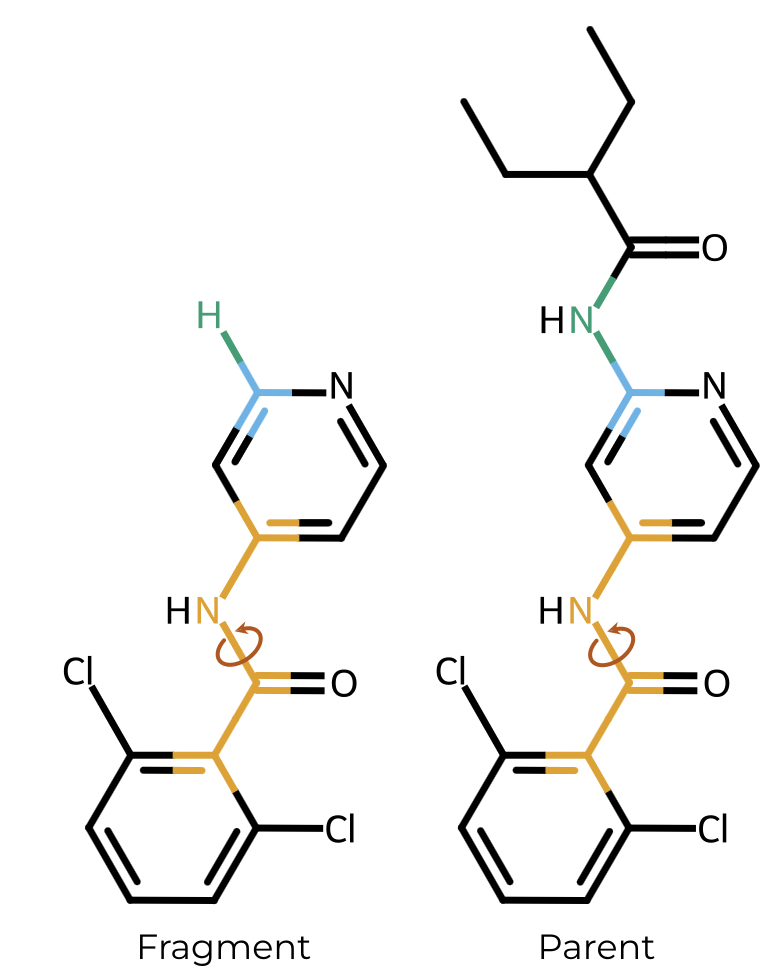
OpenFF-BespokeFit is a result of a collaboration with the Open Force Field Initiative. It facilitates the optimisation of molecule-specific force field torsion parameters against quantum mechanical, semi-empirical or machine learning potential reference data at-scale. It is compatible with the OpenFF Toolkit and QCSubmit. BespokeFit is available on GitHub, along with full documentation.
Lead developer: Josh Horton.
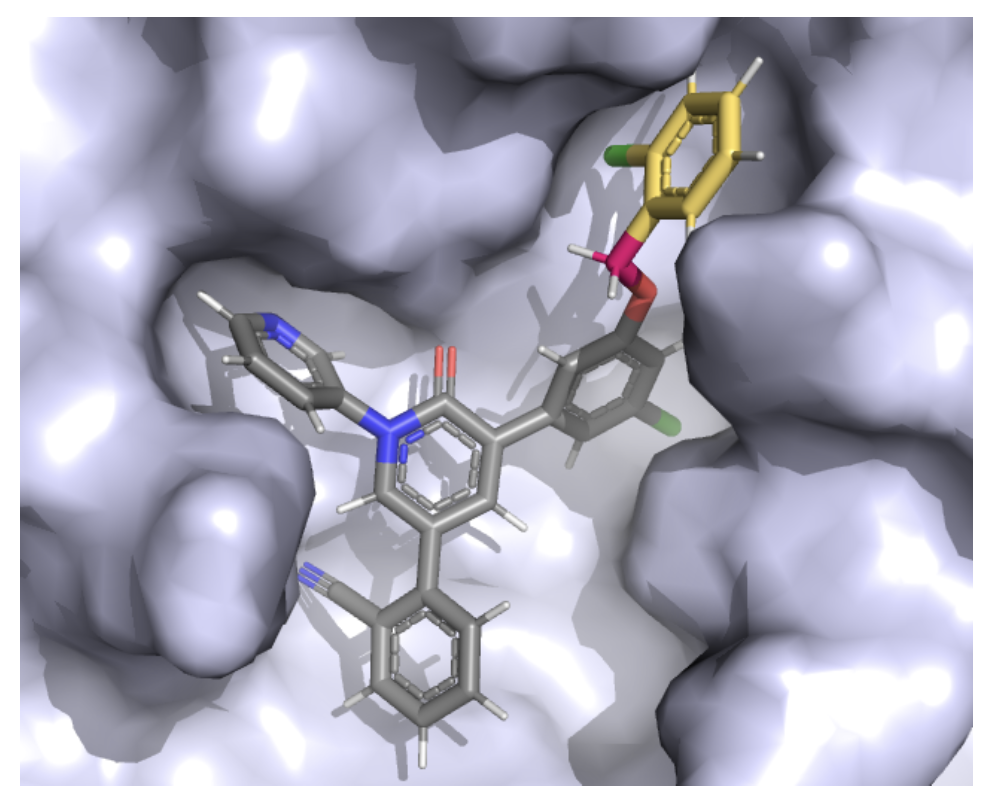
FEgrow is an open-source workflow for building congeneric series of molecules into protein binding pockets, as a stand-alone de novo design platform or for input to free energy calculations. It uses hybrid machine learning/molecular mechanics potential energy functions for building, and gnina for scoring. The latest version includes active learning and dask interfaces for automated de novo design. FEgrow is available on GitHub with a full set of tutorials and documentation. Lead developers: Mat Bieniek, Ben Cree.
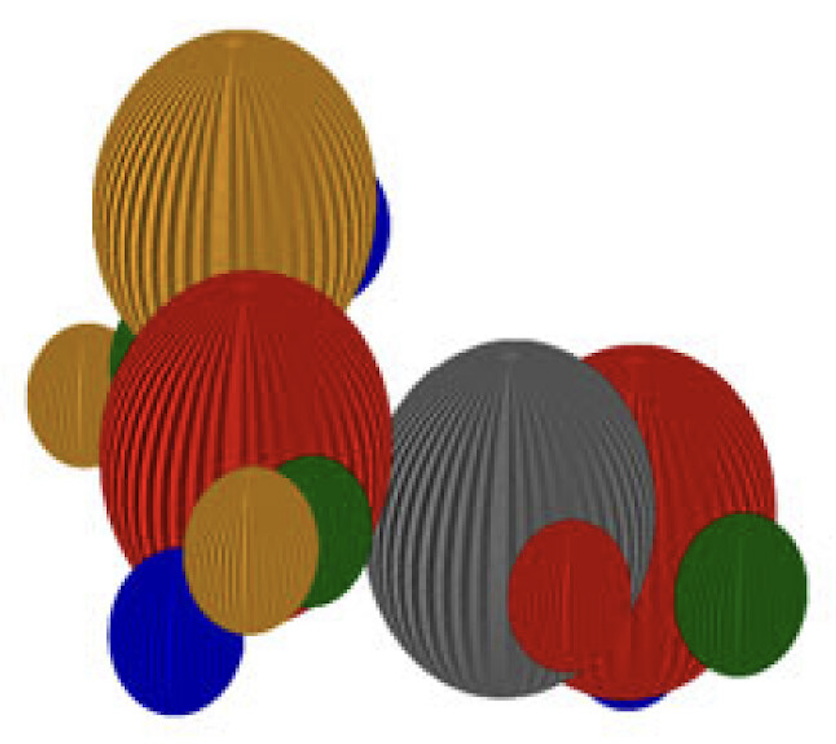
Ligand-based virtual screening aims to reduce the cost and duration of drug discovery campaigns. Shape similarity can be used to screen large databases, with the goal of predicting potential new hits by comparing to molecules with known favourable properties. RGMolSA is an alignment-free and mesh-free surface-based molecular shape descriptor derived from the mathematical theory of Riemannian geometry. In KQMolSA, Kähler quantisation is used to further transform the metric into a Hermitian matrix descriptor of shape. RGMolSA and KQMolSA are available on GitHub here and here. Developer: Rachael Pirie.

QUBEKit enables the automated derivation of molecular mechanics force field parameters directly from quantum mechanics. Incorporated techniques include atoms-in-molecule analysis for charge, Lennard-Jones and virtual site parameters, the modified Seminario method for bond and angle parameters, and automated torsional scans for dihedral parameters. QUBEKit is available on github. Lead developers: Josh Horton, Chris Ringrose.
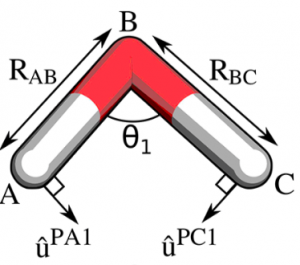
Software for the automated derivation of harmonic bond and angle force constants for molecular mechanics force fields is available on github.
Author: Alice Allen (University of Cambridge)
Allen AEA, Payne MC, Cole DJ. Journal of Chemical Theory and Computation 2018, 14(1), 274-281