Computer-Aided Drug Design
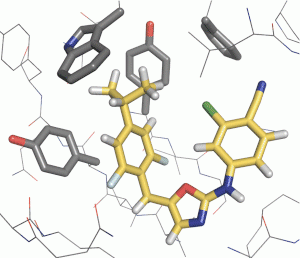
Optimisation of protein-ligand binding affinity is a central goal of small molecule drug discovery. A wide range of computational methods have been developed for this purpose, but free energy methods are a particularly attractive option, because they provide a rigorous theoretical means to compute changes in the free energy of binding, limited only by the completeness of the sampling and the accuracy of the force field. Much of our work (see below) centres around developing force fields to improve the accuracy of free energy calculations, but we also contribute to areas including de novo drug design, workflows for absolute binding free energy calculations, and shape similarity measures. We work with the Newcastle University Centres for Cancer and Rare Disease to apply these methods prospectively, and further efforts include the design of inhibitors of the aurora A kinase – TPX2 protein-protein interaction and the SARS-CoV-2 NSP13 helicase.
Open Force Field
We are passionate about open science, and proud to collaborate with the Open Force Field Initiative, and the wider Open Molecular Software Foundation. You can read a status report about the work of Open Force Field here. We have contributed to of one of the mainline force fields (‘Sage‘), and led the development of the OpenFF-BespokeFit package for fitting molecule-specific torsion parameters to high-level reference data. As an example proof-of-concept work, we experimented with using the Open Force Field software stack to replace the Lennard-Jones functional form with a more flexible double exponential function. We are continuing to work with the community to further drive forward force field accuracy and ease-of-use.
Wider Force Field Design Efforts
Quantum mechanical calculations provide an accurate description of the electronic structure of matter, but the sampling of conformational dynamics of biomolecules is beyond the reach of these techniques. Our first forays into force field design revolved around methods to extract parameters of the classical force field directly from quantum mechanics. Outcomes included the modified Seminario method for bond and angle parameter derivation from the Hessian matrix, which is now used by Open Force Field, and non-bonded parameter assignment from atoms-in-molecule analysis. We developed the QUBEKit software package to automate the entire force field assignment process, and benchmarked the accuracy on protein-ligand binding free energies. More recently, we are pleased to collaborate with the Csányi group in the development of GAP, ACE and MACE-OFF machine learning potentials for organic molecules, and with Kuano, through a knowledge transfer partnership, for the development of graph neural network based charge models.
Large-scale Density Functional Theory
Recent progress in linear-scaling density functional theory (DFT) software allows electronic structure calculations of systems comprising many thousands of atoms to be performed on a routine basis, allowing access to typical length-scales in many biomolecules. Some of the areas in the biological sciences where DFT can play an important role include the energetics of chemical reactions in enzymes, binding of small molecules by metalloproteins, and the parameterisation of model Hamiltonians to describe energy transfer in photosynthetic light-harvesting complexes.