I feel privileged to be leading such a nice and brilliant team! Finally, here is a our first group photo, at the Christmas party last Thursday with our Immunology colleagues. From right to left, Inma, Maninder, Kat and myself. Merry Christmas!!


I feel privileged to be leading such a nice and brilliant team! Finally, here is a our first group photo, at the Christmas party last Thursday with our Immunology colleagues. From right to left, Inma, Maninder, Kat and myself. Merry Christmas!!
Introns were originally thought to be ‘junk DNA’ without function but accumulating evidence has shown that they can have important functions in the regulation of gene expression. In humans and other mammals, introns can be extraordinarily large and together they account for the majority of the sequence in human protein-coding loci. However, little is known about their structural variation in human populations and the potential functional impact of this genomic variation. To address this, we have studied how copy number variants (CNVs) differentially affect exonic and intronic sequences of protein-coding genes. Using five different CNV maps, we found that CNV gains and losses are consistently underrepresented in coding regions. However, we found purely intronic losses in protein-coding genes more frequently than expected by chance, even in essential genes. Following a phylogenetic approach, we dissected how CNV losses differentially affect genes depending on their evolutionary age. Evolutionarily young genes frequently overlap with deletions that partially or entirely eliminate their coding sequence, while in evolutionary ancient genes the losses of intronic DNA are the most frequent CNV type. A detailed characterisation of these events showed that the loss of intronic sequence can be associated with significant differences in gene length and expression levels in the population. In summary, we show that genomic variation is shaping gene evolution in different ways depending on the age and function of genes. CNVs affecting introns can exert an important role in maintaining the variability of gene expression in human populations, a variability that could be related with human adaptation.
Nice press release about our latest paper.
You can visualize the chromatin states at the UCSC browser trackhub and read the full paper here:
Automatic identification of informative regions with epigenomic changes associated to hematopoiesis, by Enrique Carrillo-de-Santa-Pau, David Juan, Vera Pancaldi, Felipe Were, Ignacio Martin-Subero, Daniel Rico and Alfonso Valencia on behalf of The BLUEPRINT Consortium
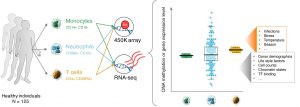
Sex is a fundamental but frequently overlooked biological characteristic of humans and model organisms that affects immune responses. We are developing integrative bioinformatics approaches to interrogate publicly available transcriptomics and epigenomics datasets to delineate the sex- determined molecular mechanisms that modulate the immune system.
We aim to generate models of the sex- and cell-specific gene regulatory networks for the major blood cell types where data is available. We will study how these sex-specific networks derived from healthy cells are influenced by infections and other disease conditions. We will develop new bioinformatics tools to integrate the sex-specific transcriptional programs with diverse sources of epigenomics information to identify the distinct chromatin configurations that underlie the different immune responses in men and women.
These results will provide the necessary framework to understand the molecular differences in men and women in response to infections, autoimmune disease and in immunodeficiencies. This will provide new insights underlying disease pathogenesis and facilitate personalised therapy for men and women.
A postdoc position funded by the Wellcome Trust is available if you will like to participate in this project! The successful candidate will the study sex-specific regulation in immune cells (data integration from publicly available transcriptomics and epigenomics databases).
Informal queries before applying are highly encouraged! Please email me if you are interested: . Click here to apply.
I am really happy to announce that I have been awarded with a Wellcome Trust Seed Award in Science – I am really honoured to be the first Early Career PI at the Newcastle Faculty of Medical Sciences to get this prestigious award!
We will study Sex-dependent gene regulation in immune cells, mining publicly available transcriptomics and epigenomics datasets. The goal is to understand how the “same” immune cell type can show different gene expression patterns in men and women, and how these differences can explain their different immune responses. We are based at the Immunology, Inflammation and Immunotherapy research theme of the Institute of Cellular Medicine and will collaborate with my experimental colleagues to experimentally explore the relevance of our findings in different disease contexts.
A post-doc position funded by the associated funding is available, please contact me if you are interested! 😉
The order in which DNA is copied during the cell division process reflects the evolutionary history of living beings: the oldest genes are copied first, and afterwards the genes that appeared later
The authors propose an original model that would explain how regions of the genome that are copied later on facilitate the birth of new genes with specific functions in tissues and organs
Press release of the work by David Juan et al (Biology Open 2013) carried out with Alfonso Valencia at CNIO.
Source: A CNIO study recreates the history of life through the genome | cnio.es
Assortativity, a property used in network analysis, helps to identify the proteins that ensure the correct folding of DNA inside the nucleus
The new perspective allows the integration of different kinds of Big Data, unravelling the 3D configuration of the genome
Last year’s press release of the work by Vera Pancaldi et al (Genome Biology 2016) carried out with Alfonso Valencia at CNIO.
Source: From happiness on Twitter to DNA organisation | cnio.es
A new paper from the group is out!! A great collaboration within the BLUEPRINT Consortium, fantastic effort leaded by Simone Ecker (former PhD student at CNIO, now postdoc with Stephan Beck at UCL).
http://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1156-8