In the latest of our series on the Early Career Researchers of ICaMB we asked Dr Yulia Yuzenkova to tell us about her research and the route that took her from a PhD in Moscow to being awarded a Royal Society University Research Fellowship in Newcastle.

- Dr Yulia Yuzenkova
My early training was in Moscow, firstly as an undergraduate at Moscow State University and then for a PhD from the Russian Academy of Sciences. However, during my PhD I moved to the USA to study at the Waksman Institute (Rutgers University) with Prof Konstantin Severinov, where I was also a postdoc. My work in the laboratory was dedicated to the molecular mechanisms of inhibition of bacterial transcription by antibacterial peptides and small proteins from viruses. Transcription is the first step and critical regulatory checkpoint of gene expression. In all living organisms transcription is performed by multi-subunit RNA polymerases (RNAP). The central role of transcription in cellular metabolism and the presence of domains specific for bacteria make RNAP an obvious target for antibiotics. Yet, for decades, only one inhibitor of RNAP, rifampicin, has been used in the clinic to treat tuberculosis, while only recently, in 2011, lipiarmycin (fidaxomicin) was approved to eradicate Clostridium difficile. Two is a very small number, but this indicates that there is a good probability of finding new drugs that also work by targetting RNAP. Apart from their clinical significance, antibiotics and inhibitors of transcription in general have been proven to be very efficient molecular tools (see “RNAP details” below for more info).
During my second PostDoc in the lab of Prof Nikolay Zenkin in Newcastle, I have focused on the mechanisms controlling the fidelity of transcription. The copying of genetic information by RNAP is far from being absolutely precise, and RNAP whimsically dislikes reading some DNA sequences, resulting in ‘pauses’. RNAP is able to correct its own mistakes using a proofreading mechanism and the help of small proteins called transcript cleavage factors (explained in detail below). It seems to be important to have at least one cleavage factors; otherwise, the RNAP molecules stop, resulting in “traffic jams” as trailing molecules keep moving and bump into it.

Batch culturing of cyanobacteria
It was therefore a big surprise for me to learn that one large group of bacteria, cyanobacteria (details below), do not encode anything even remotely resembling cleavage factors. I started to look for an explanation for this extraordinary fact. Are there any factors that might compensate for the absence of cleavage factors? Is cyanobacterial RNAP so accurate and at the same time processive that it does not need them? In searching for the answer, I realised that almost nothing is known about the molecular details of transcription in cyanobacteria, and so I decided to apply for a Royal Society University Research Fellowship to try to answer these questions.
Microscopy image of membrane-stained cell
of Synechocystis sp 6803
In performing preliminary experiments for my application, I became fascinated with cyanobacteria, as they are truly amazing organisms to work with. They are the only prokaryotes that exhibit the classic circadian clock, and are the bacteria with the most complex intracellular organisation. Moreover, they are one of the most ecologically important groups on Earth. They live everywhere where sunlight is available and produce 30% of atmospheric oxygen; some can even convert inert atmospheric nitrogen into a digestible form.

The 2D projection of the membrane structure of cyanobacterium reminds me of a labyrinth
With my Royal Society University Research Fellowship I am planning to investigate the molecular details of the transcription machinery and will look for novel transcription factors that regulate this process. I am also going to test the metal requirements of cyanobacterial transcription, because metal composition of cyanobacterial cells is very different from other bacteria to suit the need of photosynthesis. Another fascinating question is how fast in the cyanobacterial cell, which is tightly packed with photosynthetic organelles, can molecules find their way through the membrane labyrinth.
The nitty-gritty science:
RNAP in detail:
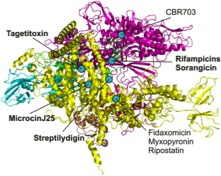
Inhibitors of RNAP as molecular tools for understanding its functions. A wide range of targets of known inhibitors is mapped on the structure of bacterial RNAP. We contributed to understanding the modes of action of inhibitors marked in bold.
Studying the antibiotics and inhibitors modes of action have helped us and other groups to discover previously unknown functions and structural domains of RNA polymerase. For example, work on rifampicin shed light on geometry of the RNA exit path, long before the crystal structure of RNAP was solved. Moreover, streptolydigin led to the discovery of the novel catalytic domain, while microcinJ25 confirmed the proposed entry channel for substrates and tagetitoxin provided insight into the mechanisms of RNAP translocation along the template.
Newly synthesised RNA participates in the proofreading in a ribozyme-like manner. This method of proofreading, probably a remnant from the distant past, is extremely slow. To accelerate proofreading (and to escape from pauses), all 3 domains of life encode non-homologous, but very similarly folded, small proteins called transcript cleavage factors. In E.coli, GreA is an example of a protein that fulfils this role. Some bacteria have several homologs of GreA (in E.coli there are at least 6). It seems to be important to have at least one, because if these cleavage factors are depleted in the cell, the RNAP molecules on the actively transcribed genes stop, obstructing transcription, but also the chromosomal replication machinery moving along the same DNA.
Cyanobacteria:
Cyanobacteria have been hailed as future photobioreactors. Indeed, when supplied with little more than tap water and light, engineered cyanobacteria can produce all sorts of compounds from sunscreen to biofuels. Cyanobacteria can also be used for environmental applications such as greenhouse gas fixing and cleaning water from industrial pollutants. These initiatives, however, are compromised by slow growth of cyanobacteria and limited knowledge of their basic biology. By putting more effort into research, the potential abilities of cyanobacteria can eventually be harnessed on the industrial level. With this we could make a giant leap towards a future “greener” economy. With a little bit of imagination, it is not hard to envisage cyanobacteria helping humanity to colonise new worlds, and to permit them to inhabit the first lunar and martial greenhouses in not so distant future.
Dr Yulia Yuzenkova’s ICaMB website: http://www.ncl.ac.uk/camb/staff/profile/yulia.yuzenkova
Royal Society University Research Fellowship: https://royalsociety.org/grants/schemes/university-research/




 In the latest of our series focussing on the Early Career Researchers (ECRs) in ICaMB, we feature
In the latest of our series focussing on the Early Career Researchers (ECRs) in ICaMB, we feature